Federal Regulations as Accelerators and Impediments to Innovation:
The TAVR Story
Haider Warraich, M.D.
Fellow, Advanced Heart Failure and Transplantation, Duke University Medical Center, Durham, North Carolina
Introduction: Medical Devices and Cardiovascular Disease Innovation
Technological innovation has seen the development not only of better pharmaceutical products, but also, increasingly, of medical devices. One of the most innovative such medical advances has been transcatheter aortic valve replacement (TAVR), which has revolutionized the management of patients with aortic stenosis, a lethal condition frequently occurring in older adults. The introduction of TAVR in the U.S., however, lagged behind dozens of countries and the procedure was not made available for commercial use for patients until several years after it had been used in other high-income countries. Such barriers affected the initial introduction of TAVR as well as the downstream innovation and development of procedural competency among U.S. operators. It has since been argued that many patients in the U.S. could have been eligible for TAVR when it was initially available in most of Europe, but several U.S.-based companies could not remain competitive given the greater regulatory thresholds they needed to surpass compared to European companies. At the same time, even as TAVR has proved to be a life-saving therapy, many cardiac devices have shown a lot of promise but ended up either providing no benefit (e.g., renal sympathetic denervation catheters1) or causing harm (bioresorbable stents2). Therefore, regulation policies frequently have to be balanced between the need to ensure patient safety and not stifling innovation or creating any unneeded delays in potentially beneficial technology reaching patients.
The medical device industry is one of the most heavily regulated industries in the U.S. Federal regulation policies, devised and implemented by the Food and Drug Administration, are first and foremost meant to ensure patient safety by preventing harmful devices from reaching the market. However, overly restrictive policies can prevent advances such as TAVR from both being developed in the U.S. and reaching American patients in a timely manner. Federal device regulations, however, do not necessarily have to be restrictive and can maintain patient safety while spurring innovation. However, the TAVR story encapsulates both ends of the spectrum and provides lessons in how such a balance can be achieved.
Aortic Stenosis
Aortic stenosis (AS) is a condition that mostly affects older individuals and involves calcification of the aortic valve, leading to obstruction of blood exiting the heart. The natural history of AS was first described by Dr Eugene Braunwald in 1958.3 Currently, Dr. Braunwald is a professor of medicine at Harvard Medical School and is the founding chairman of the Thrombolysis in Myocardial Infarction research group based in Boston. In a telephone interview, he provided some context to his initial description:
So in 1958, I was in the cath lab at the NIH. It was a very busy clinical service back then. I was working closely with a talented cardiac surgeon. Glenn Morrow, who is obviously deceased, was a great teacher and influenced me profoundly. I remember at a cath conference, before echocardiography, all we had to evaluate people by was clinical examination, which was way more detailed than it is now. This young man with aortic regurgitation, not aortic stenosis, came in.
The surgeon asked him, “What would you expect would be his prognosis in 5 to 10 years?“
“That caught me short,“ Dr. Braunwald said. “I said now that therapy is moving in this direction, we need to learn the natural history of this condition. So we began to work on this. And it turned out that we were able to put something together in aortic stenosis, but not aortic regurgitation.“ Dr. Braunwald’s description, which has since been validated, showed that as soon as patients with AS develop symptoms, they experience very high mortality. These symptoms include chest pain, heart failure and syncope.
However, unlike most cardiovascular conditions, no effective medical or pharmacologic treatment exists for severe symptomatic AS. AS is a purely mechanical pathology and its definitive treatment has been surgical aortic valve replacement (SAVR). SAVR, one of the most commonly performed cardiac surgeries in the U.S., has traditionally provided a durable and effective treatment for severe AS. However, many patients cannot receive SAVR due to the prohibitive surgical risk. Surgical risk for AVR has traditionally been assessed using a risk score derived from the Society of Thoracic Surgeons (STS) database.
The first minimally invasive device to successfully treat cardiac disease in humans was in fact developed not in a well-regulated engineering environment, but in the kitchen of a German radiologist and cardiologist, Andreas Gruentzig, in 1977. Dr. Gruentzig used a homemade balloon delivered to the coronary artery of a patient, which was then dilated to displace atherosclerotic plaque obstructing the blood vessel, using the nascent technique called cardiac catheterization. This demonstration led to the development of minimally invasive techniques such as balloon angioplasty and coronary stenting for the treatment of atherosclerotic cardiovascular disease affecting the coronary arteries. These minimally invasive procedures were equivalent and, in some ways, superior to the surgical treatment for coronary atherosclerosis, that is, coronary artery bypass grafting. A similar paradigm shift would occur in the management of AS, although it would lag behind coronary atherosclerosis management by several decades.
The Development of TAVR
The development of minimally invasive techniques to manage AS center on Alain Cribier, an instrumental figure in almost every advance in this field since the 1980s.4 A French interventional cardiologist, Dr. Cribier is widely considered the pioneer of the minimally invasive management of AS, which has today culminated in the development and widespread use of TAVR. In an email interview, he described the origins of his pioneering work:
We had a number of patients suffering from symptomatic aortic stenosis and the only possible treatment was SAVR. However, surgery was very frequently declined because of old age (age per se above 70 years was in the 1980s a usual contraindication for surgery), or bad left ventricle or cardiac and noncardiac comorbidities. For these patients, death occurred within months or after one or two years. As a possible alternative to surgery, I initiated a palliative treatment consisting of dilating the valve with a balloon catheter. This technique, called balloon aortic valvuloplasty (BAV), was first performed by our group in September 1985. The first publication in The Lancet led to an incredible enthusiasm in the medical community with thousands of inoperable patients receiving BAV in Europe and the United States until 1992.5
BAV is a relatively crude procedure involving the dilation of a balloon across the stenotic aortic valve. Although this procedure led to improvement of AS grade in some patients, it came with several major limitations. The procedure offered no mortality benefit, with AS recurring in 80% of patients at one year.6 Furthermore, the rate of serious complications was very high, with 25% of patients experiencing serious complications within the first 24 hours after BAV, and 8% experiencing cardiovascular death before discharge from the hospital.7 However, the procedure spread quickly around the world, in large part due to high complication rates from SAVR and surgery not being offered to many elderly patients, leaving them no other viable option.
Dr. Cribier’s group continued to methodically innovate, with the goal of developing a valve that could be placed through a minimally invasive approach.8 In his interview, he explained,
Developing TAVR has been a long bumpy road. … I had first to convince myself about the feasibility of using a “stented valve“ in spite of the calcific nature of the disease and the surrounding structures, the coronary arteries just above, the mitral valve and the His bundle within the interventricular septum just below. I was encouraged by the results of an autopsy study on fresh specimens of aortic stenosis that we performed in Rouen. It showed that a stent, 23 millimeters in diameter and 17 millimeters in height, could open all valves, and could be set at a distance of the surrounding structures. The concept of valvular stenting was thus validated. A valvular structure had to be inserted within the stent. I did some drawings of transcatheter valve and made a model of the device. With this in hand, and during five years, I looked for a financial partner but the experts (cardiac surgeons) of all biomedical companies you can name were steadfastly against the project, emphasizing the multiple risks of the procedure. The idea was even considered the most stupid idea ever heard. The negative opinion of experts was definitely the greatest barrier.
In 1999, Dr. Cribier formed a startup company, Percutaneous Valve Technologies (PVT), in New Jersey, with a subsidiary in Israel, to develop the first TAVR device. After animal testing, Dr. Cribier implanted this device for the first time in a human on April 16, 2002. The patient was a 57- year-old male with severe AS and heart failure, who had previously failed to benefit from BAV. The procedure was successful and Dr. Cribier later wrote: “No words can express the emotion felt by the whole team. We were witnessing a true resurrection.“ Edwards Lifesciences (Irvine, CA) subsequently bought PVT in 2004 for $125 million. The valve, nowadays referred to as the Sapien valve, developed by Dr. Cribier and subsequently acquired by Edwards, was approved for commercial use in Europe in 2007. Also in 2007, another valve, referred to as the Core Valve and subsequently acquired by Medtronic (Minneapolis, Minnesota), was approved. However, these valves were not approved in the U.S. — and even then, only to the highest-risk patients—until 2011 and 2014, respectively, four and seven years after they were commercially approved in Europe (Figure 1). In fact, the U.S. was the 42nd country to approve the Sapien device for use. By the time the Sapien device was introduced in the U.S., European cardiologists had moved on to much more advanced iterations, which had fewer complications and improved efficacy.

Innovation in the medical device sector now requires a multidisciplinary perspective. However, given the nature of the products, the U.S. also created the most robust regulator and surveillance ecosystem for monitoring the safety of these devices. Important differences exist, however, between FDA approval and the approval process in Europe, which led to the asymmetric rollout of TAVR devices on either side of the Atlantic.
U.S. FDA Device Approval Process
The FDA’s premarket approval process for devices is divided into four stages as outlined in Figure 2, and it is widely considered the most rigorous regulatory framework in the world.9 Interviews with stakeholders revealed that, given the robust resources of the FDA, much of the world relies on the U.S. regulatory framework to fully assess the safety of medical devices. Most major devices will undergo FDA evaluation, and therefore other countries look closely at these assessments even after devices have been approved by their regulatory bodies.

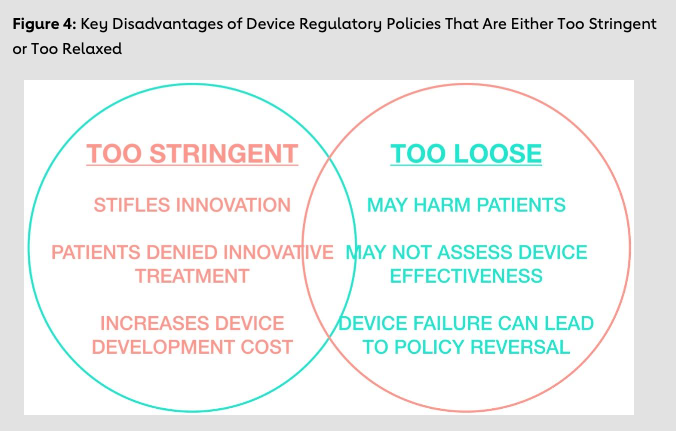
A regulatory framework that is overly onerous can have serious consequences for patient care, research and entrepreneurship. These consequences are particularly apparent when a lifesaving procedure such as TAVR is held up in the regulatory process. However, despite the delays such an environment can create, having a robust regulatory environment offers innumerable advantages, and lenient policies have led to several major regulatory failures (Figure 3).
Severe AS is a lethal condition, and in the first PARTNER trial, TAVR resulted in a 20% absolute risk reduction in death at one year when compared with patients who did not receive TAVR.10 This reduction represents some of the strongest results from any medical therapy in recent times, and therefore could have made a significant difference to patients in the U.S. who were not surgical candidates during the four years it took the U.S. to approve this device after Europe did. Second, even when the Sapien was approved, patients in other territories were already getting second- or third-generation valves that were far better than the original. Third, the earlier approval of TAVR in Europe allowed cardiologists to gain proficiency and therefore achieve better outcomes for their patients than in the U.S. Although TAVR is a unique outlier in the degree of benefit it is able to provide, this example is important because of its implications for patients.

A regulatory environment that is costly and time-consuming can stifle innovation, as was also borne out in the TAVR story. One example is Direct Flow Medical Inc. (Santa Rosa, California), a company that marketed a TAVR device that had already received CE Mark approval but could not fund the large clinical studies necessary for approval, particularly when competing against established giants such as Edwards and Medtronic. The company had been trying to secure funding from a Chinese entity, which eventually backed out, leading to the immediate shutdown of the company and 250 layoffs.
An environment where innovation and product development are too challenging produces myriad intangible effects. Although the U.S. has significant advantages given the size of the market, the presence of the most cutting-edge clinicians and researchers, and a population that is determined to receive the latest treatments possible, perceived regulatory hurdles have led to most first-in-human implantations being moved outside the U.S. Furthermore, such perceived hurdles have led to many in the public championing “Right to Try“ legislation, which seeks to circumvent the traditional regulatory pathways to allow unproven therapies to reach critically ill patients. Although this legislation failed to pass the U.S. House in 2018, the continued perception of delays in device approval will lead to these proposals gaining popularity and could jeopardize the greater regulatory landscape in the U.S.
However, loosening regulations is not the answer, given that these devices can cause considerable harm if not fully vetted, TAVR included. One particular device called Portico, developed by St. Jude Medical, offers an instructive example. The Portico device had previously received CE Mark approval in Europe in 2012, and the IDE study was initiated in the U.S. However, the trial was halted prematurely because of an incidental finding that a subset of patients experienced abnormal movement of the valve leaflets, suggesting a thrombus might have been obstructing leaflet motion.11 This abnormality went away with anticoagulation, strengthening the suspicion that this obstruction was related to leaflet thrombosis, a previously unrecognized complication of TAVR. These developments led to both the IDE trial being halted and cessation of the device’s use in Europe. Although this finding was subsequently found to potentially be unrelated to thrombus, and the CE Mark was reinstated for Portico, this example shows the additional surveillance of these devices in the U.S. regulatory process, which is co- opted by other regulatory bodies in lieu of having similar safeguards themselves. However, results from the IDE study for Portico, which has now resumed, have not been published.
Traditional CE Mark Approval
Traditionally, the European regulatory environment has been less stringent, less time consuming, and less resource-intensive for medical device approval.12 In Europe, medical devices are evaluated by one of more than 75 notifying bodies (NBs) that are private entities approved by the EU member states (Figure 2). These NBs can grant CE Mark approval; however, this process has several limitations. Companies can seek approval from any NB they deem to be the most lenient. In addition, variation exists in the standard practices, as does a lack of transparency and conflict of interest given that the company directly pays the NB for its services. A regional ethics committee provides ethical approval. The European regulatory standard is lower than that in the U.S. and requires only that the manufacturer show the device is safe and performs consistently with its marketed use. A demonstration of the device’s effectiveness in treating specific disease indications, usually through randomized controlled clinical trials with hard clinical outcomes, is not required. With regards to the Sapien valve, it received CE Mark approval based on case series and single-arm registries rather than randomized clinical trial data as required by the U.S. FDA.
The traditional CE Mark approval process has been attractive for device developers, leading to Europe becoming the site for many first-in-human device implantations and subsequent approvals. However, this lax process has led to several high-profile incidents of devices causing preventable harm to the patients they reached. Notable failures of European regulation include faulty breast implants and gastric balloons. These incidents drove European regulators to revise their regulatory process, which, according to many in the device sector, has become even more stringent than traditional FDA regulations. Some fear these regulations will be too onerous and will lead to a reduction in the number of manufacturers in Europe and an increase in costs related to regulatory approval. This pendulum shift is evident within the TAVR world as well given that the Medtronic Core Valve Evolut Pro, a newer TAVR device, received FDA approval in March 2017, whereas CE Mark approval wasn’t obtained until July 2017.
As mentioned above, CE Mark approval does not require devices to demonstrate efficacy, which is another potentially problematic feature of this regulatory process. Failure to incorporate lack of efficacy testing was most strongly demonstrated in the approval of renal sympathetic denervation catheters for patients with “refractory hypertension.“ Although catheters used to perform this procedure obtained CE Mark approval and were performed widely across the U.S., the data used for this approval and subsequent use did not include a “sham procedure.“ When a sham-procedure controlled study was performed, the device was shown to be no better than the placebo effect of the procedure.13 This example shows the immense need for efficacy data to be part of the approval process given that without it, knowing whether a device provides any actual benefit to the patient is impossible, which can greatly alter the risk-benefit and cost calculus that might drive subsequent clinical use of the device.
As the CE Mark approval process evolves, some innovators, like Dr. Cribier, are encouraged:
The European Commission has recently published new regulation for medical devices with the main goal of strengthening patient safety before placing new devices on the market. In consequence, there is no doubt that the regulatory environment will be more stringent. The applicable standards of medical devices will be reinforced and the new regulation will at last harmonize the rules all over Europe. In this regard, the new regulation is moving in the right direction to improve the safety of the devices and provide more transparency for patients.
Innovation in Device Approval: Insights From FDA Leadership
Leadership at the FDA took important lessons from the delays in achieving approval for the original Sapien valve.14 The FDA is accelerating the use of real-world data to allow for a swifter and less laborious regulatory ecosystem. This step involves the development of the National Evaluation System for health Technology (NEST). In an exclusive interview with the FDA’s Changfu Wu, Ph.D., lead reviewer at the Structural Heart Devices Branch, and Bram Zuckerman, M.D., director of the Division of Cardiovascular Devices, Center for Devices and Radiological Health, innovative efforts underway at FDA were highlighted:
The NEST system would generate evidence and support analysis across the medical device life cycle by strategically and systematically leveraging real-world evidence derived from data that is collected during routine patient care. It would apply advanced analytics to data tailored to the unique data needs and innovation cycles of medical devices. The FDA envisions the system would link and synthesize data from different sources across the medical device landscape, including clinical registries, electronic health records, medical billing claims, data transmitted from devices and other digital sources. As part of the foundation for the system, the FDA is establishing a Universal Device Identifier system and promoting its incorporation into electronic health information, facilitating the development of national and international device registries, and developing new methods to collect, analyze, and communicate knowledge about medical devices globally.
The result of lessons learned from the TAVR story have already led to tangible reforms. According to Dr. Wu, “On June 5th, 2017, FDA became the first regulatory body in the world to approve the most recent iteration of the Sapien valve, the Sapien 3, to treat high-risk patients whose surgically placed aortic or mitral bioprosthetic valves were old and worn out.“
These reforms became possible through a novel collaboration between the FDA, device companies and academic cardiovascular societies. Dr. Wu continues:
Some 100,000 patients have received TAVR since FDA’s first approval in 2011, including more than 600 patients for what were then off-label, valve-in-valve uses. FDA relied on real-world evidence to evaluate the benefits and risks of this off-label use — such as the safety of the procedure, the function of the valve, and the improvement of patient symptoms — to approve the new indication for Sapien 3. … The expanded indication approval was made possible using real-world evidence, in this case by the Transcatheter Valve Therapy (TVT) Registry, a partnership of the American College of Cardiology and the Society of Thoracic Surgeons. The TVT registry collects clinical data on the performance of transcatheter valve replacement procedures performed in the U.S. once a product goes to market — including both on-label and off-label uses — making it possible, under certain circumstances, to accumulate more data faster, without the need for costly and time-consuming formal clinical trials.
The overarching goal of newer changes is to shorten the time required for a device to receive approval, but also to enhance post-approval surveillance to assess device efficacy and effectiveness in clinical practice using real-time data gathered from NEST. Such an innovation, spearheaded by the FDA, can help bridge a divide between the need to expedite approvals to drive innovation and the need to maintain patient safety. To achieve this lofty goal, another key program is the Medical Device Epidemiology Network Initiative (MDEpiNet), a public-private partnership that is part of the Epidemiology Research Program at the FDA’s Center for Devices and Radiological Health. The goal of this program is to enhance the capability of real-world data sources to answer questions with regulatory value without always having to rely on laborious and expensive clinical trials. MDEpiNet collaboratively provides tools including study design for distributed network-based research collaborations, advanced analytical overall methods such as multilevel analyses (hospital, surgeon, patient), advanced analytical methods for confounding adjustment (propensity scores, instrumental variables, cross-design syntheses and Bayesian methods), and tools to help strengthen relationships and stakeholder development. By bringing together academic medical centers, manufacturers, payers, registries and patients, MDEpiNet provides a platform for collaboration, which could be used to address challenges across the spectrum of the device-regulation ecosystem.
FDA regulations can play an important role by requiring long-term data on the effectiveness of new innovations. One case that is of particular importance is that of bioresorbable stents.15 Coronary stents are placed in patients with atherosclerotic plaque to restore blood flow to the heart. Manufacturers developed a new type of stent, which over time could get resorbed. This innovative product was meant to solve some of the long-term issues with stents, such as in-stent restenosis and the need for multiple stents to be placed in patients over time. Although initial results were extremely promising, more long-term data revealed that patients receiving the stent experienced a high rate of late stent thrombosis, a potentially fatal complication. This issue led to the manufacturer, Abbott, shutting down the program.
The FDA is evolving its approach to device approval and, according to Dr. Wu, “is recalibrating its balance of a device’s benefits and risks prior to market against assuring that patients have faster access to the treatments they need.“ By establishing NEST, the goal is to have more effective post-market surveillance but to shorten the upfront burdens for market entry:
The agency plans to seek more flexible authorities to place mitigations as conditions of a device being used — like more control over a device’s label including new patient risks and more consistent user training. The agency will leverage some of our existing authorities under the 21st Century Cures Act of 2016 and the FDA Reauthorization Act of 2017 whenever possible as well as leverage our regulations and identify funding to meet our goals.
The need to provide faster access for patients must be heavily counterbalanced by the need to protect patients. When regulators fail to provide safeguards, harmful devices can reach the market, which can seriously damage consumer confidence. Within cardiology, several drug- eluting stents were approved in Europe in the absence of robust safety and efficacy data. These cautionary examples demonstrate that regulation can protect innovation by ensuring harmful or ineffective products don’t reach the market. Early detection of a potentially harmful product can also prevent the manufacturer from investing too heavily in a product that ultimately might not be able to recoup those costs. A summary of pitfalls of regulations that are either too stringent or too lenient is provided in Figure 4. However, as this figure shows, an overlap exists where regulations can both accelerate innovation and provide safety, and this intersection needs to be better studied and implemented.
The Way Forward: Building a Regulatory Bridge Between Safety and Innovation
TAVR represents one of the most significant leaps in therapy for cardiovascular disease in recent years, leading to the development of a subspecialty within cardiology focused on implantation of these devices. TAVR has also opened the door for innovations to treat other cardiac valves (mitral, tricuspid and pulmonary valves), using minimally invasive techniques. Note that the TAVR story is an outlier because of how beneficial the procedure turned out to be, and simply using the challenges faced during the TAVR regulatory approval process to make inferences about the entire process might be a stretch. U.S. regulators have, however, already learned important lessons.
The TAVR regulatory process in the U.S. was undesirably long, yet it generated the strongest, most robust data about the efficacy and safety of TAVR devices. Although Europe might appear to have made a better decision through swifter approval, that judgment is heavily colored by the hindsight knowledge that TAVR is, in fact, efficacious and safe. The European CE Mark designation was granted to TAVR in the absence of any randomized, controlled clinical trial data, and lax European standards have consistently exposed patient populations there to devices that are either harmful or futile. EU members who have recognized these drawbacks have designed newer regulations far more stringent than those of the U.S. Therefore, the traditional European regulatory pathway may not be the optimal source after which to model an ideal regulatory ecosystem.
Several possible opportunities to streamline the regulatory process in the U.S. have been proposed. One would be to streamline approvals by ethical board reviews and payers. Another issue that prevents the U.S. from being a site for early human testing of devices is litigation requirements, which need to be balanced with the need to introduce these devices earlier to U.S. patients.
Since the arrival of TAVR, the FDA has moved in a direction that is encouraging innovation. A key legislative piece was the 21st Century Cures Act, which President Obama signed into law in December 2016. This act expands data sources beyond clinical trials to help the decision process for drug and device approval. These sources include data from observational registries, the electronic health record and so on. Better understanding of these data sources may improve their utility to monitor the safety and efficacy of new devices. Having improved surveillance systems will push the evaluation of these devices into the post-marketing phase after introduction rather than delay entry of devices into the market. Therefore, the future for device development is bright in the U.S. The key will be to create regulations that serve as accelerators of innovations but that don’t harm patients or expose the health system to ineffective and expensive devices.
Endnotes
- D.L. Bhatt et al. A controlled trial of renal denervation for resistant hypertension. N Engl J Med 2014;370:1393- 401.
- Cassese S, Byrne RA, Ndrepepa G, et al. Everolimus-eluting bioresorbable vascular scaffolds versus everolimus- eluting metallic stents: a meta-analysis of randomised controlled trials. Lancet 2016;387:537-44.
- Morrow AG, Sharp EH, Braunwald E. Congenital aortic stenosis; clinical and hemodynamic findings, surgical technic, and results of operation. Circulation 1958;18:1091-104.
- Cribier A. Development of transcatheter aortic valve implantation (TAVI): a 20-year odyssey. Arch Cardiovasc Dis 2012;105:146-52.
- Hara H, Pedersen WR, Ladich E, et al. Percutaneous balloon aortic valvuloplasty revisited: time for a renaissance? Circulation 2007;115:e334-8.
- Cassese et al., “Everolimus-Eluting Bioresorbable.“
-
-
Driving Innovation
-
Innovations in Cardiovascular Health
-
The Role of Physicians in Driving Innovation
-
The Role of Patient Groups in Driving Innovation
-
Clinical Innovations in Cardiovascular Health
-
What Drives Innovation in CV Health?
-
The Rise of Academic and Contract Research Orgs
-
Federal Regulations as Accelerators
-
Reimbursement Models
-
Consumer Technology
-
Training Cross-Disciplinary Innovators
-
Conclusion
