The Rise of Academic and Contract Research Organizations
Alexander Fanaroff, M.D.
Perelman School of Medicine, University of Pennsylvania
Introduction
Since the 1960s, the age-standardized death rate from cardiovascular disease has decreased by nearly 60% in the U.S.1 Approximately half of that decline has been attributed to better control of cardiovascular risk factors, including lipids and blood pressure, and the other half has been attributed to therapies for treating acute myocardial infarction (MI), secondary prevention following MI, and pharmacologic and device therapies for heart failure.2 A critical catalyst for the development and dissemination of these therapies was the advent of large, multinational clinical trials. These “mega-trials,“ which enroll tens of thousands of patients with cardiovascular disease, enable researchers to demonstrate relatively small differences in important but relatively rare events, such as death following MI. Though these events are rare, and treatments are only modestly effective, the results of large cardiovascular clinical trials have critical public health implications because of the high prevalence of cardiovascular disease in the population.
Academic research organizations (AROs) and contract research organizations (CROs) have played a key role in the evolution of modern cardiovascular trials. Both AROs and CROs are hired by research sponsors, including pharmaceutical companies and device manufacturers, as well as governmental agencies such as the National Institutes of Health, to assist the sponsor with the conduct of clinical research. AROs are affiliated with universities and are generally not- for-profit; CROs are generally for-profit corporations. From the 1950s to the 1990s, academic medical centers worked with pharmaceutical companies and regulatory bodies (the U.S. Food and Drug Administration and European Medicines Agency) to develop the basic design elements of the modern cardiovascular clinical trial. These elements included key features of how to operationalize global cardiovascular “mega-trials“ as well as their analysis. AROs grew up and their operational infrastructures developed to support these trials. Furthermore, grand alliances were formed among the regional AROs to support global enrollment. Once the basic pattern and tactics for these large trials were well established, CROs were able to scale them, and commoditized these processes, facilitating global clinical research on a large scale.
Since these early days, however, clinical trials have grown increasingly and unsustainably complex and costly.3-6 As a result, industry has begun to abandon many promising pharmaceutical compounds and medical devices due to the cost and inherent economic risk associated with development. But such challenges also become the grounds for evolution in the industry. Currently, the development of pragmatic clinical trial design has generated excitement.7-9 AROs have once again begun to pioneer novel and simpler clinical-trial designs. These studies often use data found within the electronic health records (EHR) of health systems to foster more efficient patient identification, data collection and longitudinal patient follow- up,10,11 and frequently partner with patients as active study participants. Within this evolving new landscape, the ARO once again is finding its way as a source of innovation within the clinical trial world.
In this chapter, we will describe the historical development of the modern clinical-trial industry, conditions in the 1980s and 1990s that led to the rise of CROs, innovations developed by CROs that facilitated large-scale clinical research, and innovations developed by AROs to offset the rising costs and decreasing enrollment of clinical research, and project future directions for continued innovation by AROs and CROs.
The Rise of Academic and Contract Research Organizations
Birth of the Modern Clinical Trial and the Academic Research Organization
The story of the rise of AROs and CROs is intricately tied to the development of the modern clinical-trial industry — as of 2017, a $63 billion business that involves AROs, CROs, site- management organizations, hospital systems, pharmaceutical companies, biotechnology companies, information technology companies and government agencies. The modern clinical- trial industry would not exist without AROs and CROs, nor would AROs and CROs exist without the modern clinical trial industry. A series of scientific, regulatory and social changes in the 20th century changed the level of evidence required for clinicians, patients and regulators to accept a new therapy, creating a need for large-scale clinical research, which AROs and CROs helped fill.
Though clinical trials were conducted as early as the 18th century, the basic structure and statistical underpinnings of modern randomized clinical trials were established in the early 1950s by Sir Austin Bradford Hill.12-14 By the latter part of the decade, groups of investigators were conducting multicenter clinical trials with patients with cancer.15 Despite these advances, most medical and regulatory decision-making was guided by expert opinion, or by the advertisements and salesmen of a rapidly growing pharmaceutical industry.16 The Food, Drug, and Cosmetic Act of 1938 required pharmaceutical companies to show evidence of their drugs’ safety prior to marketing, but many products were tested only in short-duration animal studies prior to widespread use in humans. Physicians believed their clinical judgment was sacrosanct.
After a series of harmful and ineffective drugs were marketed — including, most notably, thalidomide — Congress passed the Kefauver-Harris Act in 1962. The Kefauver-Harris Act included the requirement that new drugs demonstrate safety and efficacy prior to being marketed in “adequate and well-controlled investigations“ conducted by “experts qualified by scientific training.“17 This legislation both set the regulatory ground rules for pharmaceutical development in the U.S. and the world, and established the norm that scientific testing, rather than expert opinion, was the standard by which drugs (or, eventually, other therapies) would be judged.18 These norms were fostered in the 1940s by the contribution of penicillin to U.S. victory in World War II, and reinforced in the 1950s by a financial commitment to medical research. Between 1950 and 1968, funding for medical and scientific research grew 15-fold, from $161 million to $2.5 billion, and the National Institutes of Health both opened its research hospital and began funding clinical research through its peer-reviewed, extramural grants system.15
By 1970, in a report in the Journal of the American Medical Association on the ongoing Coronary Drug Project, investigators expressed the rationale for large clinical trials evaluating cardiovascular outcomes:
[Physicians] know that susceptibility to first episodes of premature CHD is directly related to serum levels of cholesterol, and low-density and very-low-density lipoproteins. They are also aware that elevated serum lipids-lipoproteins frequently can be reduced long-term by available drugs. However, they lack the answers to key questions about these pharmaceutical agents: Do they prevent recurrent episodes of CHD and prolong life? … Are they reasonably safe, in long-term usage?20
The Coronary Drug Project was the largest cardiovascular clinical trial yet performed. Sponsored by the NIH and coordinated by the University of Maryland School of Medicine, the trial enrolled 8,341 patients with prior MI at 53 U.S. sites over four years.21 After the Coronary Drug Project, clinical trials studying cardiovascular outcomes became more prevalent, but most clinical trials in the 1970s and early 1980s were small-scale studies with fewer than 1,000 patients, conducted by individual academic health centers or small consortia (fewer than 15 sites),22-24 and most prospective patient-oriented research published in major medical journals remained single- center, sometimes single-arm, studies enrolling fewer than 200 patients with short-term or mechanistic outcomes.25-27 One exception to this rule, the Anturane Reinfarction Trial, an industry-coordinated randomized trial of the antithrombotic agent sulfinpyrazone in 1,558 patients at 26 sites in the U.S. and Canada, illustrates how many of the rules and rituals governing clinical trials had not yet been codified.28 The initial publication in the New England Journal of Medicine claimed that sulfinpyrazone reduced the risk of sudden cardiac death in the first six months following acute myocardial infarction,29 and the Ciba-Geigy Corp., which manufactured sulfinpyrazone and conducted the trial, sought FDA approval for that claim. However, the study was criticized for the use of non-standardized cause-of-death definitions and reliance on an as-treated rather than an intention-to-treat analysis, and the FDA ultimately declined to approve sulfinpyrazone for this indication.30
A series of changes in the late 1970s and 1980s set the stage for the major pharmaceutical company-funded clinical trials that largely started in the mid-1980s and continue to represent the standard for cardiovascular clinical trials today (Figure 1).
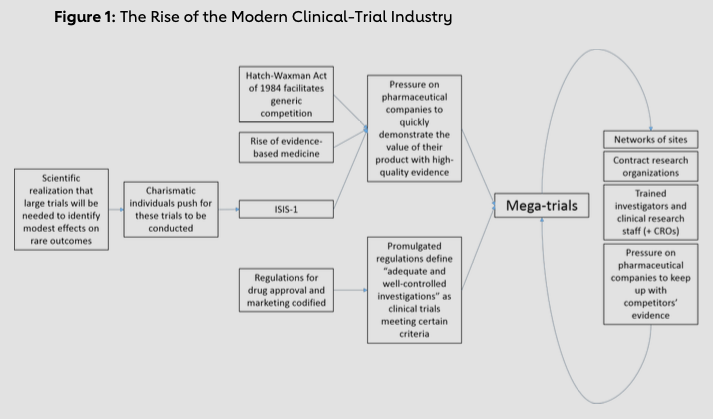
First, many of the rules and rituals of clinical trials were codified as a result of the Drug Efficacy Study. In this program, largely conducted from 1966 to 1969, the National Academy of Sciences reviewed the evidence of efficacy for all drugs approved prior to 1962; the FDA planned to remove from the market drugs ruled ineffective.15 FDA reviewers published hundreds of critiques of study methodology, and eventually scientific opinion coalesced around the idea that clinical trials should have a valid hypothesis, a prospectively defined endpoint and a pre-specified analytic plan.15 Changes in FDA practices in the late 1970s, and regulations in the 1980s and 1990s, further increased collaboration between the pharmaceutical industry and regulators; regulators routinely provided input into the design of clinical trials, clarifying the rules and rituals of the clinical-trial process.
Second, the Drug Price Competition and Patent Term Restoration Act (informally known as the Hatch-Waxman Act), passed by Congress in 1984, provided a financial incentive for pharmaceutical companies to conduct large studies designed to move products quickly through the regulatory pipeline. Though the Food, Drug, and Cosmetic Act of 1938 provided a process for the approval of generic drugs, in practice, the FDA approved few generic drugs; in 1983, for example, only 35% of top-selling branded drugs had generic competition, and generic drugs only captured 13% of the market for pharmaceuticals.33 Without the time pressure of an expiring patent, pharmaceutical companies could afford to conduct the small studies needed to achieve narrow regulatory approval while relying on expert opinion to slowly increase uptake of newly approved agents. The Hatch-Waxman Act prevented the FDA from seeking information from companies seeking to produce generic drugs, other than information on how the company planned to manufacture the drug and a study demonstrating bioequivalence with the approved drug. This legislation opened the door to generic medications, increasing the pressure on pharmaceutical companies to move drugs quickly from pre-clinical development to widespread uptake. Large clinical trials powered to detect differences in key clinical outcomes were one way to convince a global audience of cardiologists of a new drug’s value and create a “blockbuster“ drug.
Third, cardiologists were primed to accept the importance of clinical-trial evidence to guide practice by the rise of evidence-based medicine in the 1980s.34 Some of these changes were driven by charismatic and visionary individuals. In the 1970s, Sir Richard Peto, working at Oxford University in the UK, developed many of the statistical methods used to analyze clinical-trial results and was among the first to note that detecting modest treatment effects on the incidence of rare events would require the inclusion of thousands of patients.35 Groups of investigators in Europe and the U.S. coalesced around these ideas. The first international mega- trial was the International Study of Infarct Survival (ISIS)-1 study, conducted by the Oxford Clinical Trials Unit. Investigators examining the effect of beta blockers on infarct size in animal models approached Peto with a question about how large a study would need to be to detect a difference in cardiovascular outcomes with beta blockers versus placebo among post-MI patients, and Peto found that it would take many thousands of patients. The investigators began a clinical trial in the UK with the financial support of Imperial Chemical Industries, the manufacturer of atenolol and the forerunner of AstraZeneca. Working without a budget, the investigators and the company engaged academic cardiologists and Imperial Chemical Industries’ subsidiaries in other European countries. The academic cardiologists enrolled patients in other countries, and the subsidiaries provided drugs to the investigators. Ultimately, ISIS randomized 16,027 MI patients at 245 hospitals in 14 countries to atenolol or placebo, making it the first CV mega-trial.36 ISIS-1 demonstrated significantly lower seven-day and one- year mortality among patients randomized to atenolol than among those randomized to placebo.
The ISIS investigators followed up with large randomized controlled trials enrolling 17,187 patients and 41,299 patients, respectively.37,38 The Italian investigators who collaborated on ISIS-1 spun off their own group, Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardio (GISSI), which began conducting similarly sized randomized controlled trials.39-41 These studies demonstrated the power of large clinical trials and also developed a critical mass of site investigators and trial operational personnel to conduct large-scale clinical research. The success of these trials captured the attention of other pharmaceutical companies, and the era of CV mega-trials was born.
In the U.S., the Thrombolysis and Angioplasty in Myocardial Infarction group pivoted from conducting smaller, mechanistic trials in the late 1980s and 1990s42 to coordinating the Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries (GUSTO) clinical trial, which randomized 41,021 MI patients at 1,081 hospitals in 15 countries to four different thrombolytic strategies.43 The resources and site networks developed for GUSTO ultimately evolved into the Duke Clinical Research Institute. Other academic medical centers with experience in clinical-trial management, including the Oxford Clinical Trials Unit, the Thrombolysis in Myocardial Infarction (TIMI) study group in Boston, the Population Health Research Institute in Canada and the Cleveland Clinic Coordinating Center for Clinical Research would replicate this model to varying degrees, becoming AROs in the 1990s. AROs offer various services to pharmaceutical companies and other clinical trial sponsors: thought leadership, site management (usually within the same country or region), statistical analysis, data management and site monitoring.
Commodification and Commercialization: The Rise of Contract Research Organizations
By the mid-1990s, the rules and rituals of clinical trials were well codified, and AROs, with their networks of sites, investigators and research staff with experience managing large-scale clinical research, had formed partnerships with the pharmaceutical industry to lead the conduct of some of these trials. Other trials were managed by pharmaceutical companies’ in-house clinical-research teams. For a number of reasons, however, the pharmaceutical industry was not satisfied with this arrangement, which created a niche in which CROs grew and thrived. The first two CROs, Quintiles (now IQVIA) and Parexel, were founded in 1982 — Quintiles as a biostatistics consulting firm and Parexel as a regulatory consulting firm. Throughout the 1980s, CROs were relatively small players in the cardiovascular clinical-research enterprise: Academic health centers captured 80% of industry-sponsored clinical research in 1988.44 By the early 2000s, academic health centers captured only 30% of industry-sponsored research, with the remainder going to CROs.45 CRO revenues increased from $1 billion in 1992 to $41 billion in 2016.46 A number of factors — including pharmaceutical companies’ drive for efficiency, globalization of research, opportunities to develop and deploy new technology and changes in the structure of clinical research — led to the growth of CROs.
The major driver was pharmaceutical companies’ need for efficiency, related both to speed and costs. The Hatch-Waxman Act’s facilitation of generic competition for new brand-name medications reduced the amount of money pharmaceutical companies could make off of each drug and pressured them to complete development and testing quickly to maximize the amount of time they could market a drug before patent exclusivity expired. Automated screening protocols developed by pharmaceutical companies produced a rapidly growing list of candidate drugs for testing; between 1990 and 2000, the number of drugs in phase 1 trials increased from 386 to 1,512.47 Some studies estimate that delays in completing clinical trials cost pharmaceutical companies over $1 million/day in lost revenues.48,49 With clinical-trial and FDA- approval procedures well understood, drug testing could be reduced to a commodity, the speed and expense with which a trial was conducted evaluated based on its contribution to the bottom line.50
CROs offered a number of advantages in speed and efficiency compared with AROs and pharmaceutical companies’ in-house clinical-research teams. The academic culture of AROs prized the independence of investigators and broad, challenging scientific questions without certain answers, not research-for-hire based on narrow questions with a sure answer. This academic culture of independence was also relatively uncompromising with regard to the investigators’ right and responsibility to publish clinical-trial results promptly and without bias; pharmaceutical companies’ motives sometimes failed to align with this ethos.51 Pharmaceutical companies also had to pay a “Dean’s Tax“ to work with AROs, because universities used industry-funded clinical research to support money-losing divisions and departments. Unlike university-based AROs, CROs did not have an academic culture or alternative missions that taxed revenue from projects.
AROs also tended to view each clinical trial as an artisanal product, an ethos that led to trials frequently being more complex, having more data collection and taking longer and costing more than had the only goal been to most efficiently determine the efficacy and safety of a given candidate therapy. But by the 1990s, the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) had standardized regulatory requirements for drug development between countries.52 Regulatory audits were also formalized, and groups tasked with adhering to regulatory requirements at pharmaceutical companies grew larger and more powerful. CROs could use this standard template and offer pharmaceutical companies more efficient and standard procedures that would meet these regulatory requirements.
Furthermore, CROs had economies of scale working for them. AROs, being part of academic institutions, generally were conservative in their expansion of staff. As such, they tended to remain small. Similarly, in-house clinical-research operations within pharmaceutical companies were always struggling to accommodate the changing demand for research-personnel capacity that fluctuated based on the number of drugs they had undergoing active testing.53 When capacity was reached, pharmaceutical companies outsourced research-personnel needs, protecting them from the expense of maintaining employees and capacity needed for peak clinical-research capability when research volume was in a trough. Biotechnology startups, which arose in the 1980s, had fewer employees than traditional pharmaceutical companies and no appreciable clinical-research capacity, thus providing CROs with more sources of business. By contrast, CROs were backed by investors and could maintain capacity to conduct hundreds of clinical trials at the same time, without worrying about ebbs and flows related to the drug- development cycle at an individual pharmaceutical company.
Globalization also fueled CROs’ rise. Conducting research in low- and middle-income countries was cheaper than doing so in higher-income countries, creating another advantage for research operations with global capacity. Pharmaceutical companies’ national divisions were managed as separate entities, preventing efficient collaboration on multinational clinical trials, and AROs were geographically confined by the location of their university, but CROs had no such limitations. When CROs in separate countries merged, as they frequently did through the 1990s, the single entity instantaneously had site networks, clinical-trial-management experience and regulatory expertise in both countries. CROs quickly became capable of running transnational clinical trials, which AROs and pharmaceutical companies’ in-house research operations could not match.
The rise of CROs was also facilitated by emerging technology. With the advent of inexpensive personal computing, solving data-management problems became the key to clinical trial operations. However, leveraging emerging technology to more efficiently conduct clinical research required an upfront investment in software development. AROs were slow to invest in disruptive and cost-saving clinical-research technology, such as electronic data capture or image-interpretation software. By contrast, CROs conducted hundreds or thousands of clinical trials at the same time and could apply economies of scale to the development of these technologies. Some also supported the development of novel, cost-saving technologies by selling them to AROs and other CROs.
Finally, one of the major competitive advantages of the AROs was trust, but this advantage became less important over time. The academic and independent nature of AROs promoted a belief that the answers they derived from a clinical trial were more “believable“ than those run by a for-hire CRO or run internally by a pharmaceutical company. However, as the FDA and others developed better auditing of clinical trials, honesty was compelled, and universities’ reputations for intellectual honesty were no longer critical. Additionally, major journals, such as the Journal of the American Medical Association, Lancet, and the New England Journal of Medicine, once required that the results from trials be verified by independent academic statistical teams; however, by the 2000s, competition among the journals drove a relaxation of these requirements.
As CROs captured more and more of the market for conducting clinical trials, they were further able to apply economies of scale. Different firms competed aggressively with each other, driving down costs, with some CROs even taking on some of the financial risks of drug development. CROs and pharmaceutical companies formed preferred partnerships, with discounts based on volume. In some cases, CROs, with their lower costs and sometimes less-skilled workforces, enabled pharmaceutical companies to better match the needs of a given clinical trial to the amount spent on it: Clinical trials of a novel drug for a new indication were managed in-house or by AROs by more skilled (and more expensive) clinical-research teams, whereas late-phase clinical trials of a “me-too“ drug could be outsourced to a CRO.16
Rising Costs: A Crisis in Clinical Research and an Opportunity for Innovation
Despite competition and innovation by CROs and AROs, by the mid-2000s, the rising cost of clinical research became a problem, especially in the U.S. Between 1991 and 2003, clinical research cost per new drug increased more than fourfold after adjusting for inflation, from $104 million in 1991 to $467 million in 2003.55 By 2013, the cost to conduct a large phase 3 or 4 clinical trial alone was estimated at $400 million.56 Higher costs are driven by a number of interrelated factors, which have been reviewed in detail elsewhere,57 including complex trial procedures,58,59 diminishing site-level trial participation (with a resultant need to increase site payments to encourage participation),60 decreased perception of the need for clinical research by patients and physicians, and a complex regulatory environment (including the need for multiple site- level IRBs to approve a research protocol, specialized contracting with each site, inefficiencies in serious adverse-event monitoring, and aggressive auditing and monitoring). Pharmaceutical companies (and the CROs they hire to conduct clinical research) have responded to these pressures by offshoring an increasing proportion of their clinical trials from the U.S. to less developed, less regulated, less costly regions of the world: North American patients typically comprise fewer than 20% of patients enrolled in both NIH- and industry-funded clinical trials.61,62 Though this practice may have reduced the cost of generating regulatory-ready clinical-trial data, it results in difficulty for regulators to assess the effect new drugs may have in the U.S. and raises questions about monitoring at non-U.S. sites.64 Furthermore, studies to answer many critical public health questions will not be funded by industry sources, and the increasing costs of clinical research price out governmental funding sources.
The next disruption: pragmatic clinical trials
AROs, working with government agencies, have led in the design of innovative, streamlined approaches to clinical-trial conduct, including risk-based monitoring, focused adverse-event reporting and use of real-world data sources and quality-improvement registries as platforms for clinical trials.64 Broadly, these methods have been termed pragmatic clinical trials. These novel approaches have been used in government-funded clinical trials but have not been as common in industry-funded research.
In the U.S., the Duke Clinical Research Institute led the NIH-funded Study of Access Site for Enhancement of Percutaneous Coronary Intervention (SAFE-PCI) for Women clinical trial, which used baseline patient data collected for a quality-improvement registry for the purposes of a clinical trial comparing radial with femoral access in patients undergoing PCI.65 A Swedish ARO, the Uppsala Clinical Research Institute, has leveraged comprehensive cardiovascular registries that incorporate long-term follow-up to perform clinical trials of anticoagulation in patients undergoing PCI, thrombus aspiration in patients with ST-segment elevation MI, and oxygen in patients with MI, without collecting additional trial-specific data.66-68 These registry-based randomized clinical trials can be conducted at approximately one-tenth the cost of a similarly sized traditional trial.69,70
AROs have also led the development and testing of methods for using administrative claims data and patient reporting to capture outcomes in randomized clinical trials and other multi- center clinical-research efforts.71,72 In ARTEMIS (Affordability and Real-world Antiplatelet Treatment Effectiveness After Myocardial Infarction Study), ARO investigators developed a process for capturing clinical endpoint data from hospital bills and patient reporting, validating events by central adjudication.73
AROs have also worked with government agencies to transform real-world data into clinical trial-ready datasets,74 building platforms for future pragmatic clinical trials that may not require central adjudication. One such effort, PCORNet, the National Patient-Centered Clinical Research Network, will enable electronic health record data to be captured for prospective clinical trials75 The first PCORNet-enabled trial, ADAPTABLE (Aspirin Dosing: A Patient-centric Trial Assessing Benefits and Long-Term Effectiveness), has begun enrolling patients to test the efficacy and safety of two different doses of aspirin. 76 AROs have also worked with the FDA to develop methods for using the Sentinel Initiative, a distributed database of commercially insured patients, to capture research-quality data; Sentinel IMPACT-AFib, the first clinical trial to use this data source, will enroll 80,000 patients with atrial fibrillation to test a quality- improvement initiative aimed at increasing the uptake of oral anticoagulation.77 AROs’ thought leaders and statistical expertise have also been critical to repurposing administrative data for post-marketing surveillance of medical devices, lowering barriers to regulatory approval while maintaining safety.78
AROs and academic health centers have pioneered innovations such as video informed consent and virtual clinical trials, enabling clinical trials to reach patients that may not participate in traditional clinical research.79-81 They have furthered the broad-scale implementation of novel clinical-trial designs for health-services intervention and quality improvement, such as cluster randomized and stepped-wedge clinical trials, in which health systems or hospitals rather than patients are randomized.82,83 AROs’ relationships with patients and patient groups have also been critical in creating a role for patient advocacy in research design.84,85
The Future of Academic and Contract Research Organizations — Cooperation?
In 2018, AROs and CROs are best equipped to serve different roles. AROs are best positioned to focus on thought leadership, both in identifying critical clinical questions and designing new clinical-research paradigms; in conventional clinical trials, AROs cannot compete with CROs’ greater efficiency in more labor-intensive trial operations, such as site monitoring and data management. Many conventional clinical trials now involve partnerships between AROs and CROs, where AROs provide thought leadership and manage processes that address publication requirements (including independent statistical analysis) and regulations for independent academic oversight (such as oversight of data safety and monitoring boards and steering committees), and CROs focus on trial execution, site monitoring and data management.86 This “best-of-both-worlds“ approach maintains academic independence and enables efficient trial conduct, but may leave AROs ill-suited to develop trainees well versed in clinical-trial operations and design. Collaboration between AROs may help reduce the disparities of scale between AROs and CROs,87 though AROs will always be smaller.
When cardiovascular clinical trials matured in the latter half of the 20th century, academic medical centers played a key role in establishing the rituals and processes, and later developed into AROs. Once the ground rules were established, CROs developed economies of scale, turning clinical research into a commodity. With changes in the funding and regulatory environment, innovation in clinical trial methods were necessary, and AROs again became essential to developing these new methods. Once the new methods are more established, pharmaceutical companies will likely seek to use them, and CROs will again seek to commoditize them for the conduct of large-scale clinical research. By then, new crises will arise, along with new opportunities, and AROs will again be called on to innovate.
Acknowledgments
We thank the people who shared their experience and expertise, including Eric Peterson (DCRI), Pete DiBattiste (Janssen Pharmaceuticals), Rob Califf (DCRI, FDA), Rory Collins (Clinical Trials Unit, Oxford University), and Josef von Rickenbach (Parxel).
Endnotes
- H.K. Weir et al., “Heart Disease and Cancer Deaths — Trends and Projections in the United States, 1969–2020,“ Preventing Chronic Disease 13 (2016); e157.
- E.S. Ford et al., “Explaining the Decrease in U.S. Deaths From Coronary Disease, 1980–2000,“ New England Journal of Medicine 356 (2007): 2388-2398.
- R.M. Califf and R.A. Harrington, “American Industry and the U.S. Cardiovascular Clinical Research Enterprise,“ Journal of the American College of Cardiology 58 (2011); 677-680.
- K.A. Getz, “Protocol Design Trends and Their Effect on Clinical Trial Performance,“ RAJ Pharma (2008):315-316.
- K.A. Getz and R.A. Campo, “Trial Watch: Trends in Clinical Trial Design Complexity,“ Nature Reviews Drug Discovery 16 (2017): 307.
- N.S. Sung et al., “Central Challenges Facing the National Clinical Research Enterprise,“ JAMA 289 (2003): 1278- 1287.
- Z.J. Eapen et al., “The Imperative of Overcoming Barriers to the Conduct of Large, Simple Trials,“ JAMA 311 (2014): 1397-1398.
- Z.J. Eapen et al., “Rescuing Clinical Trials in the United States and Beyond: A Call for Action,“ American Heart Journal 165 (2013): 837-847.
- C.M. O’Connor et al., “Improving Heart Failure Therapeutics Development in the United States: The Heart Failure Collaboratory,“ Journal of the American College of Cardiology 71 (2018): 443-453.
- W.S. Jones et al., “The Changing Landscape of Randomized Clinical Trials in Cardiovascular Disease,“ Journal of the American College of Cardiology 68 (2016): 1898-1907.
- C.S. James, S.V. Rao and C.B. Granger, “Registry-Based Randomized Clinical Trials — a New Clinical Trial Paradigm,“ Nature Reviews Cardiology 12 (2015): 312-316.
- S.J. Pocock, “The Historical Development of Clinical Trials,“ Clinical Trials: A Practical Approach, New York: John Wiley & Sons; 1983: 14-27.
- A.B. Hill, “The Clinical Trial,“ New England Journal of Medicine 247 (1952): 113-119.
- A.B. Hill, “The Clinical Trial,“ British Medical Bulletin 7 (1951); 278-282.
- S.W. Junod, “FDA and Clinical Drug Trials: A Short History,“ About FDA; 2018.
- M. Shuchman. “Commercializing Clinical Trials — Risks and Benefits of the CRO Boom,“ New England Journal of Medicine 357 (2007):1365.
-
-
Driving Innovation
-
Innovations in Cardiovascular Health
-
The Role of Physicians in Driving Innovation
-
The Role of Patient Groups in Driving Innovation
-
Clinical Innovations in Cardiovascular Health
-
What Drives Innovation in CV Health?
-
The Rise of Academic and Contract Research Orgs
-
Federal Regulations as Accelerators
-
Reimbursement Models
-
Consumer Technology
-
Training Cross-Disciplinary Innovators
-
Conclusion
